Frequently Asked Questions (FAQs)
Question 1: What are the requirements of the query sequence for calculating a CAI?
The input requirements are described in the tutorial. Basically, the server checks whether the query sequence is in fasta format, whether it is a DNA or RNA sequence (i.e. it contains only A, C, T, G or U), whether the length of the query is divisible by three and does not contain a stop codon between the sequence.
Question 2: Which codons are used to calculate the CAIs?
CAI calculation uses only codons that have at least one synonymous codon. For example, in the standard genetic code, as the codon TGG is the only one that codifies for Tryptophan, it is not used to calculate the CAI.
Question 3: Can I calculate a CAI for more than one sequence?
Yes, you can introduce more than one sequence in FASTA format to calculate CAI.
Question 4: Can I calculate a CAI without a codon usage reference table?
No, the calculation of CAI always requires a codon usage reference table.
Question 5: Where can I find codon usage tables to use as a reference set?
Codon usage tables can be obtained from the Codon Usage Database. You can also construct and use your own codon usage reference tables.
Question 6: I have calculated the CAI of 3 sequences using human codon usage as a reference. The CAI of the sequences is 1) 0.535, 2) 0.856 and 3) 0.740. And the expected CAI for these sequences is 0.701 at 95% and 0.752 at 99%. How can I evaluate this result?
Expected CAI provides a direct threshold value that makes it possible to discern whether differences in the CAI value are statistically significant or merely artifacts arising from internal biases in the G+C composition and/or the amino acid composition of the query sequences. The CAI of sequence 1 is not significantly higher than expected according to its nucleotide and amino acid composition, because its CAI value is lower than both expected CAI values. For sequence 2, the CAI is significantly higher than expected at a 95% confidence level, and for sequence 3 it is significantly higher than expected at a 99% confidence level.
Question 7: I use Firefox 1.0.2 as navigator. Is there any problem to use CAIcal?
No. CAIcal has tested with the most common Internet navigators and runs well with both Windows and MacOs versions of Internet explorer 6.0, Netscape navigator, Mozilla, Safari and Firefox.
Question 8: How is the CAI along a sequence graphically represented?
You can choose a window size and window step. Using these values, the server moves a window along the query sequence and calculates the CAI for each window. If the window size and window step are 1, the weight of each codon in the reference set (the frequency of a codon divided by the highest frequency of the codons that code for the same amino acid) is represented.
Question 9: Can I graphically represent the CAI along a sequence using other programs?
Yes, you can copy the tab-delimited output and paste it into your favorite program.
Question 10: Why, in the graphical representation of the CAI along a sequence using a window size and a window step of one, are there some codons without a value or bar?
Because these codons do not have a synonymous codon and a weight for them cannot be computed. These codons depend on the genetic code chosen.
Question 11: I want to graphically represent the codon adaptation of a group of sequences that I have aligned. Do I need to use one or multiple reference sets?
If all the sequences are from the same source (i.e. they are paralogous sequences) you can use only one reference set. If sequences are from different species (i.e. they are orthologous sequences) and you want to see whether some regions are more adapted to each species, you need to use a reference set for each sequence.
Question 12: Is it important to maintain the same order of sequences between the protein alignment and the DNA sequences in the protein alignment section?
No. Protein sequences in the multialignment and DNA sequences are matched by their id, so the order is not important. The important thing is that each DNA sequence uses the same id as in the protein multialignment.
Question 13: Can I choose the window step and window length in the graphical representation of the CAI along a protein multialignment?
Yes, after submitting your protein multialignment the DNA-translated multialignment and its representation are shown. You can then re-draw the multialignment using a different window length or window step.
Question 14: How are the gaps considered in the graphical representation of the CAI along a protein multialignment?
For each codon position in the protein multialignment translated to DNA, the mean value of the weights of each codon is calculated. Gaps are considered as codons with a weight of 0. Positions with gaps therefore appear as positions with a poor codon adaptation.
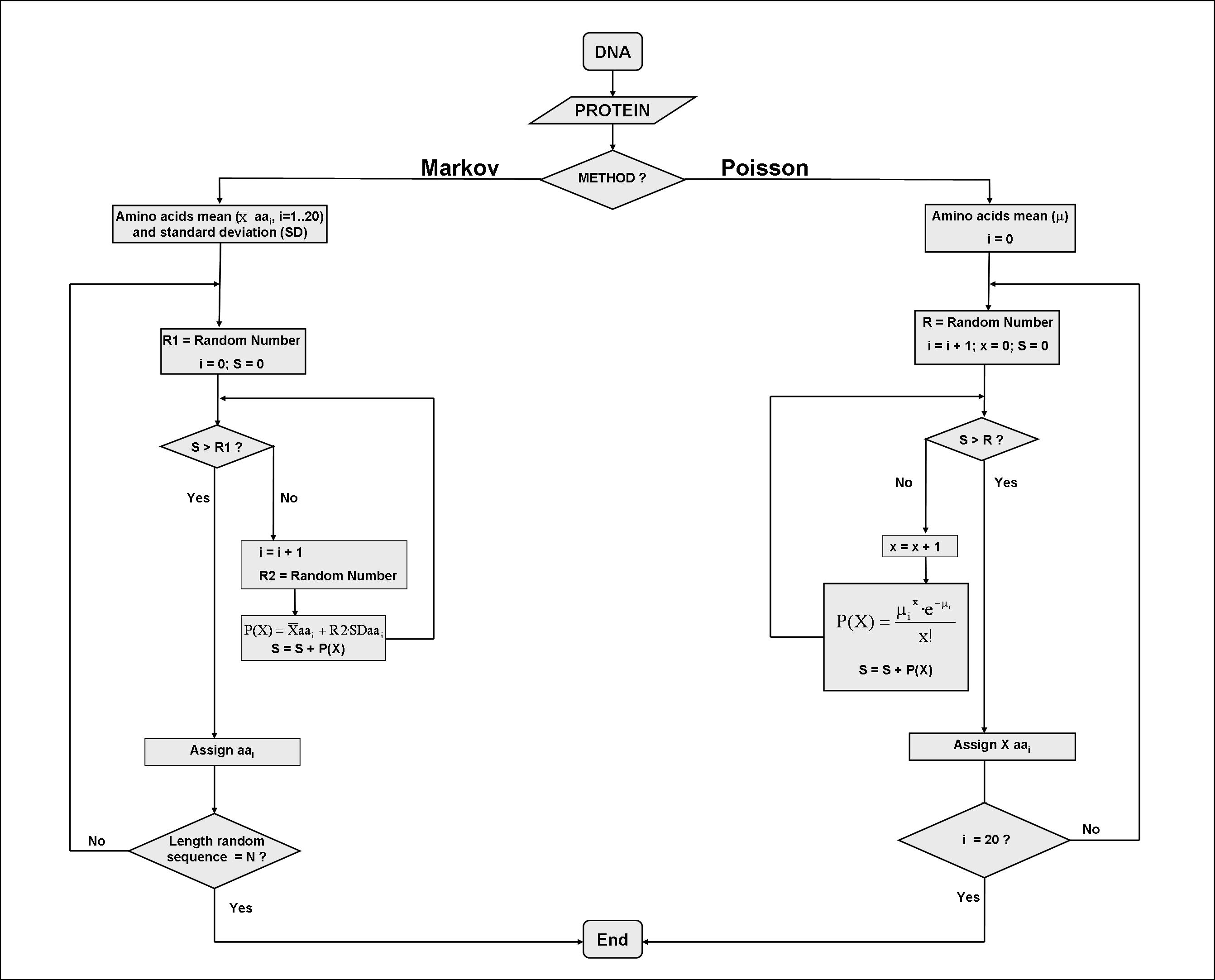
Question 15: What are the differences between the Markov and Poisson methods for estimating the expected CAI value?
Both methods are similar in the sense that, using the amino acid composition of the query sequences and random generated numbers, they generate a series of amino acid sequences that are compositionally equivalent to the query. These sequences are then back-translated to DNA using the mean G+C content of the query. The CAI of these random-generated sequences are then used to estimate an expected value of CAI. Both methods provide similar results. The differences between the two methods can be seen in the following diagram:
The Markov method is a Markov Model of order 0. This means that the probability of an amino acid to occupy a position in a protein is independent of the other sites. The Markov method generates the random sequences by adding one amino acid each time to a generated amino acid sequence, since this sequence has the desired length. The amino acid is chosen randomly using the amino acid frequencies of the query sequences and a random number. The Poisson method, on the other hand, assumes that the number of times that each amino acid is used in a protein follows a Poisson distribution. The amino acid frequencies of the query sequences multiplied by the length of the sequence to be generated are the expected values (called h) of the number of times each amino acid is used in a protein. From these expected values, the probability of finding each amino acid 0, 1, 2, 3, 4, .., n times is calculated from the expression p(n) = e(-h) hn / n! From these probabilities and a random number, the number of each amino acid in a random generated sequence is calculated.
Question 16: What are the significance level and coverage in the estimation of an expected CAI
Given a confidence limit and a percentage of the population (also called coverage) chosen by the user, an expected value of CAI is estimated using an upper one-sided tolerance interval for a normal distribution. A tolerance interval is a way to determine a range that, with a given confidence level, will contain a certain percentage of the population. In our case, the upper limit represents the value that is not exceeded by the specified fraction of the randomly generated sequences with the chosen confidence limit.
Question 17:What is the meaning of the Kolmogorov-Smirnov test in the estimation of the expected CAI?
The expected value of CAI is estimated from the mean and standard deviation of the CAI of the randomly generated sequences using a tolerance interval based on a normal distribution. To estimate this expected value, the user has to choose two parameters: the level of significance and the percentage of the population or coverage. To check whether the CAI of the randomly generated sequences follow a normal distribution, a Kolmogorov-Smirnov test is made.
Question 18: What is the meaning of the chi-square test in the results page of the estimation of an expected CAI?
To generate the random sequences, the server uses the amino acid frequencies and G+C content of the sequences from the query. The server assumes that these frequencies are representative of all the sequences of the query. To check this assumption, a chi-square test is conducted to compare the goodness-of-fit between the amino acid frequencies or G+C content of each sequence of the query and their mean values. The server shows the percentage of the query sequences that pass the test, i.e. the percentage of query sequences that are represented by the mean values. If this percentage is too low, this means that the amino acid composition or G+C content of the query is too heterogeneous. To solve this problem, we recommend estimating different expected values for compositionally different sequences.
Question 19: Is it possible to calculate the eCAI value one by one for many sequences instead of a set of sequences at one time?
The v1.1 of the source code and the graphical user interface distribution provide such output. Using the -s option, potential users can decide to calculate an unique eCAI value for a set of sequences (-s n) or to calculate, one by one, an eCAI value for each sequence(-s y).
Question 20: I suggest the authors to integrate the method used to analyze the difference between CAI and eCAI into their server.
The calculation of CAI and the estimation of the expected value are performed by different programs and appear in different response pages of the CAIcal/E-CAI server. However, this possibility is very easy to implement in a simple perl script using the local version of the E-CAI program. We have included an exemple Perl script called example.pl as an example of how implement this case.
Question 21: Is there a Windows version of the local version of E-CAI?
The local version of the E-CAI program is written in Perl. Perl is an interpreted programming language that does not need to create an executable file for every operating system or computer architecture. This is a great advantage of this kind of programming languages. Users only need the source code of the script and a Perl interpreter in order to execute any Perl script. Windows users only need therefore to install a Perl interpreter prior to execute the source code of the E-CAI local version. In the download section of the server we include the source code of the E-CAI local version and the link of a free Perl interpreter.
Question 22: Which are the differences between the 1.0 and 1.1 local versions?
The only difference between both version is that 1.1 version includes now the -s option (see Question 19).
Question 23: Is there any difference between the CAI calculation through the web server and the local version?
No, both the web server written in PHP and the stand alone application written in perl return the same results. Click here to see an example.
Question 24: Have you ever compared the values of CAI calculated in CAIcal with other similar servers?
Yes, we have calculated the CAI of more than a thousand of DNA sequences using CAIcal and using CAIcalculator2 (Wu et al., 2005). Though, CAIcalculator2 uses the original algorithm proposed by Sharp and Li (1987) and CAIcal uses this algorithm with a few modifications proposed by Xia (2007), the results from both servers are comparable. Click here to see results from this test (sequences and reference set are also available to repeat the test).
Question 25: Where can I get a complete documentation about the source code of the local version?
The source code includes several informative comments about its structure. Click here to get additional information.
Question 26: Which are the differences between the 1.1 and 1.2 local versions?
We have fixed minor bugs, such as the graphical interface did not permit to change the genetic code, and included more comments in the source code.
Question 27: Which are the differences between the 1.2 and 1.3 local versions?
We have refined the coding style and included more comments in the source code.
Question 28: What are the differences between the Markov and Poisson methods for estimating the expected RCDI value?
Both methods are similar in the sense that, using the amino acid composition of the query sequences and random generated numbers, they generate a series of amino acid sequences that are compositionally equivalent to the query. These sequences are then back-translated to DNA using the mean G+C content of the query. The RCDI of these random-generated sequences are then used to estimate an expected value of RCDI. Both methods provide similar results. The differences between the two methods can be seen in the following diagram:
The Markov method is a Markov Model of order 0. This means that the probability of an amino acid to occupy a position in a protein is independent of the other sites. The Markov method generates the random sequences by adding one amino acid each time to a generated amino acid sequence, since this sequence has the desired length. The amino acid is chosen randomly using the amino acid frequencies of the query sequences and a random number. The Poisson method, on the other hand, assumes that the number of times that each amino acid is used in a protein follows a Poisson distribution. The amino acid frequencies of the query sequences multiplied by the length of the sequence to be generated are the expected values (called h) of the number of times each amino acid is used in a protein. From these expected values, the probability of finding each amino acid 0, 1, 2, 3, 4, .., n times is calculated from the expression p(n) = e(-h) hn / n! From these probabilities and a random number, the number of each amino acid in a random generated sequence is calculated.
Question 29: What are the significance level and coverage in the estimation of an expected RCDI
Given a confidence limit and a percentage of the population (also called coverage) chosen by the user, an expected value of RCDI is estimated using an upper one-sided tolerance interval for a normal distribution. A tolerance interval is a way to determine a range that, with a given confidence level, will contain a certain percentage of the population. In our case, the upper limit represents the value that is not exceeded by the specified fraction of the randomly generated sequences with the chosen confidence limit.
Question 30:What is the meaning of the Kolmogorov-Smirnov test in the estimation of the expected RCDI?
The expected value of RCDI is estimated from the mean and standard deviation of the RCDI of the randomly generated sequences using a tolerance interval based on a normal distribution. To estimate this expected value, the user has to choose two parameters: the level of significance and the percentage of the population or coverage. To check whether the RCDI of the randomly generated sequences follow a normal distribution, a Kolmogorov-Smirnov test is made.
Question 31: What is the input table format?
An easy way to introduce the codon usage reference table in RCDI/eRCDI is to copy and paste the codon usage tables from Codon Usage Database (Nakamura et al., 2000). We have therefore added a link to this database (and also codon usage tables from model organisms) in the left frame of the server.
The codon usage table from the codon Usage Database format allowed in RCDI is as follows:
Fields: [triplet] [frequency: per thousand] ([number])...
We have also introduced another format as follows:
Fields: [triplet] [frequency: per thousand] ([number])...
Question 32: What does the "Insert %G+C content" option mean?
Users may define (optional) the %G+C of the random sequences. If this option is not select, the program uses the %G+C content of the input sequences.
Question 33:What are the results shown in the "gene's parameters table ?
The first and second columns contain the name of the gene and the RCDI respectivelly. Then, the frequency of each codon calculated as [(CiFa/CiFh)Ni] (where CiFa is the relative frequency of codon i for a specific amino acid in the test sequence; CiFh is the relative frequency of codon i for a specific amino acid in the reference sequence; Ni is the number of occurrences of codon i in the test sequence; and N, the total number of codons in the test sequence), and the last four columns contain the %G+C total and at each codon position.
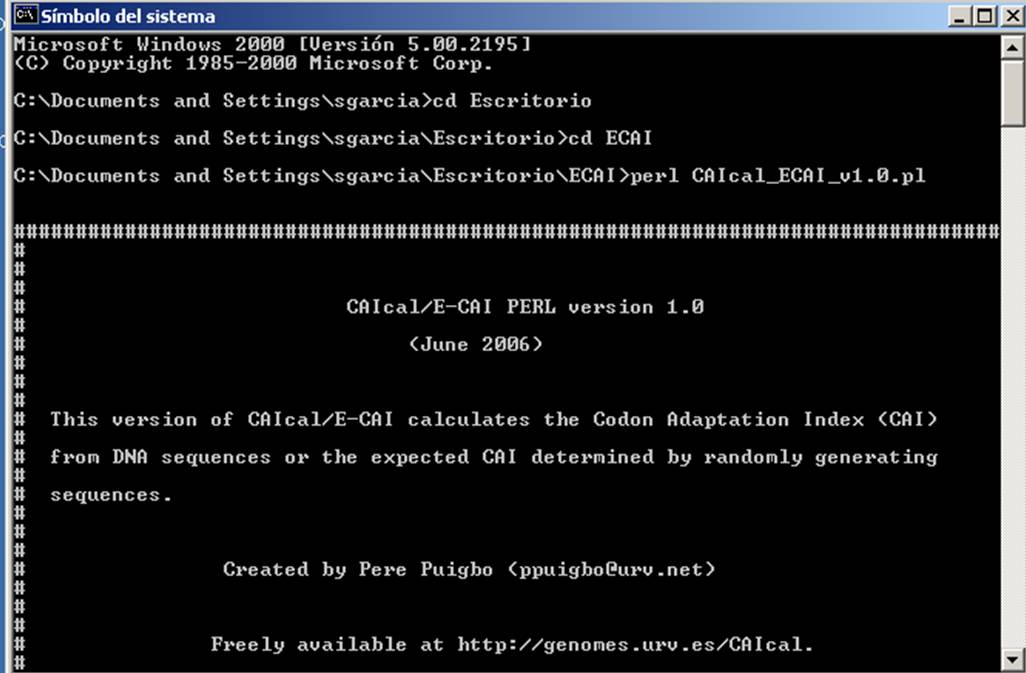
After installing the Perl interpreter, downloading and uncompressing the source code file, windows users need to open a DOS-terminal, access to the directory that contain the source code and execute the script by writing "perl CAIcal_ECAI_v*.pl" (Replace * for the appropriate version).
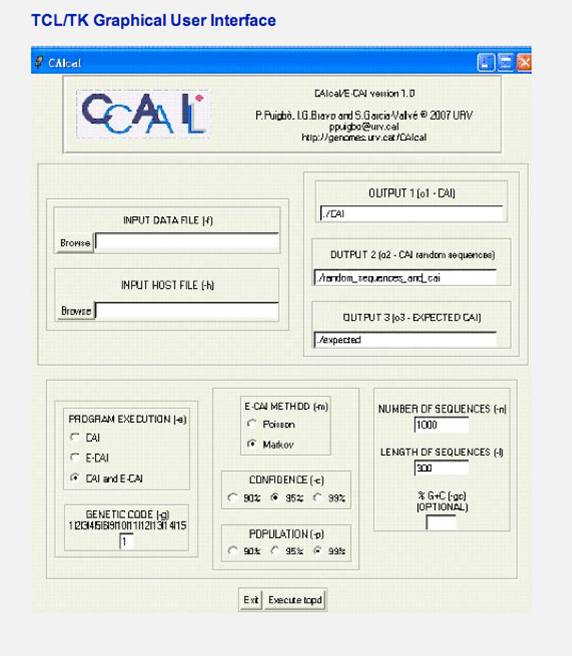
In addition, we have created a graphical interface in Tcl/Tk. Windows users only need to install the windows version of the Tcl/Tk toolkit (a link is provided in the download section), download the Tcl/Tk version of the E-CAI, unzipped and double click on the CAIcal.tcl file. The Tcl/Tk graphical interface of the E-CAI local version looks like this when run on Windows: